Usage
Python module
Quick start
Parametrization of structure with coordinates saved as supramolecular_cage.xyz with (nonbonded) topology supramolecular_cage.top (of the whole structure):
from metallicious import supramolecular_structure
cage = supramolecular_structure('supramolecular_cage.xyz',
metal_charges={'metal name 1': charge of metal 1(integer),
'metal name 2':charge of metal 2(integer),...},
topol='supramolecular_cage.top', LJ_type='uff')
cage.parametrize(out_coord='out.pdb', out_topol='out.top')
For example, for the structure ru_pd.xyz with force-field parameters saved as ru_pd.top, which consists of two metals Pd2+ and Ru2+, the input file looks like this:
from metallicious import supramolecular_structure
cage = supramolecular_structure('ru_pd.xyz', metal_charges={'Ru': 2, 'Pd':2 },
topol='ru_pd.top', LJ_type='uff')
cage.parametrize(out_coord='out.pdb', out_topol='out.top')
Initial topology file
If you don’t have a topology file, you can generate a simple force-field parametrization using General Amber Force-field (GAFF):
from metallicious import supramolecular_structure
cage = supramolecular_structure('ru_pd.xyz', metal_charges={'Ru': 2, 'Pd':2 },
LJ_type='uff')
cage.prepare_initial_topology()
cage.parametrize(out_coord='out.pdb', out_topol='out.top')
Please note that our focus was on covalent metal parametrization; only basic support for the organic molecule parametrization is available. For more robust parameterization protocols, please refer to specialized tools such as gromacs, ATB, ambertools, and charmm-gui.
Handling missing templates
The number of combination of possible ligands and metal results that inevitably you will encounter metal site for which there is no template. In such cases two solutions are possible:
1. Parametrize template
We recommend to run template parametrization on HPC/cluster as it can take some time (our experience is ~8h on 8 CPUs per template).
Specifying explicitly the metal multiplicity using the metal_charge_mult variable instead of metal_charges, will automatically inform metallicious to be ready to parametrize the new template.
from metallicious import supramolecular_structure
cage = supramolecular_structure('ru_pd.xyz', metal_charge_mult = {'Ru': (2,1), 'Pd':(2,1)}, LJ_type='uff')
cage.parametrize(out_coord='out.pdb', out_topol='out.top')
2. Truncate existing template
If an exact template is unavailable in the library, you can truncate part of an existing template. Truncation is based on the distance from the metal centre, such as 4-bonds away (“dihedral”), 3-bonds away (“angles”), or 1-bond away (“bonds”). Such a strategy is fast but results in a loss of accuracy.
For example:
from metallicious import supramolecular_structure
cage = supramolecular_structure('ru_pd.xyz', metal_charge_mult = {'Ru': (2,1), 'Pd':(2,1)}, truncation_scheme = 'dihedral', LJ_type='merz-opc')
cage.parametrize(out_coord='out.pdb', out_topol='out.top')
Input for supramolecular_structure class
The extended input list of supramolecular_structure class:
Variable |
Type |
Description |
Default |
Needs to be specified? |
|---|---|---|---|---|
filename |
str |
name of the coordination file |
none |
yes |
metal_charge_mult |
dict |
the names charges, and multiplicity of the metals in format {metal_name: (metal_charges, multiplicity)} |
none |
yes or metal_charges |
metal_charges |
dict |
the names and charges of metals in the input structure in format: {metal_name1: metal_charges1, metal1_name2: metal_charge2} |
none |
yes or metal_charge_mult |
LJ_type |
str |
name of LJ dataset used for metal paramters (uff, merz-tip3p, merz-opc3, merz-spc/e, merz-tip3p-fb, merz-opc, merz-tip4p-fb, merz-tip4-ew, zhang-tip3p, zhang-opc3, zhang-spc/e, zhang-spc/eb, zhang-tip3p-fb, zhang-opc, zhang-tip4p/2005, zhang-tip4p-d, zhang-tip4p-fb, zhang-tip4p-ew) |
none |
yes |
topol |
str |
force-field parameters file |
none |
Yes, unless later prepare_initial_topol used |
keywords |
list(str) |
autodE keywords for QM calculations |
PBE0 D3BJ def2-SVP tightOPT freq |
no |
improper_metal |
bool |
if True it will parametrize the improper dihedral involving metal |
false |
no |
donors |
list(str) |
list of atom elements with which metal forms bond |
[‘N’, ‘S’, ‘O’] |
No |
library_path |
str |
directory of template library, be default where the script is |
path to metallicious + /library |
no |
search_library |
bool |
if True, metallicious searches templates in template library,if False, it will parametrize template |
true |
no |
ff |
str |
parametrization protocol for small organic molecules (only gaff available at the moment) |
‘gaff’ |
no |
fingerprint_guess_list |
list(str) |
list of template names, which will be checked from library |
none |
no |
truncation_scheme |
str |
name of the truncation scheme |
none |
no |
covalent_cutoff |
float |
if metal-atoms smaller then cutoff it is assumed that creates bond with the metal |
3.0 |
no |
Bash command line
It is also possible to use the metallicious just from the command line. For example:
metallicious -f cage.xyz -LJ_type merz-tip3p -metal_and_charges Pd 2 -prepare_topol
For details, see:
metallicious -h
Extended list of the bash command:
Variable |
Comment |
possible input |
Default |
Required |
|---|---|---|---|---|
-h, –help |
show help message and exit |
possible input |
none |
no |
-f |
metaloorganic coordination file |
*.gro, *.pdb and other coordination formats supported by MDAnalysis |
none |
yes |
-p |
metaloorganic force-field parameters of non-bonded model |
.top, .prmtop, etc. and other supported by ParmEd |
none |
yes (unless prepare_topol specified) |
-of |
output metaloorganic structure |
.gro, .pdb and other formats supported by MDAnalysis |
out.pdb |
no |
-op |
output metaloorganic topology |
.top, .prmtop and other formats supported by ParmEd |
out.top |
no |
-metal_charges |
metal names and charges (optionally, multiplicity when parametrization needed) |
names and charges are separate by whitespace (e.g., Pd 2 Ru 2) or names, charges and multiplicities separated by spaces (e.g., Pd 2 1 Ru 2 1) |
none |
yes |
-keywords |
autodE keywords for QM calculations |
see autodE or ORCA manual |
PBE0 D3BJ def2-SVP tightOPT freq |
no |
-LJ_type |
type of parameters for Lennard-Jones parameters |
uff, merz-tip3p, merz-opc3, merz-spc/e, merz-tip3p-fb, merz-opc, merz-tip4p-fb, merz-tip4-ew, zhang-tip3p, zhang-opc3, zhang-spc/e, zhang-spc/eb, zhang-tip3p-fb, zhang-opc, zhang-tip4p/2005, zhang-tip4p-d, zhang-tip4p-fb, zhang-tip4p-ew |
uff |
no |
-truncate |
truncation scheme |
none, 3bond/dihedral, 2bond/angle, 1bond/bond |
none |
no |
-improper_metal |
calculate the improper dihedral of the metal-aromatic |
true/false |
false |
no |
-donors |
donors from the connected ligands, usually electronegative atoms, such as N, S, O, but sometimes metal is connected to carbon |
any element name separated by space |
N S O |
no |
-prepare_topol |
prepare initial topology using GAFF |
true/false |
false |
no |
-linker_topol |
linker force-field (topology) parameters, only used when prepare_topol=True |
.top, .prmtop, etc. and other formats supported by ParmEd |
none |
no |
Available parameters
Default templates
By default, metallicious contains a few templates which are commonly used in metallo-organic cages. However, more templates can be easily added using automated parametrization procedure, which is also part of metallicious.
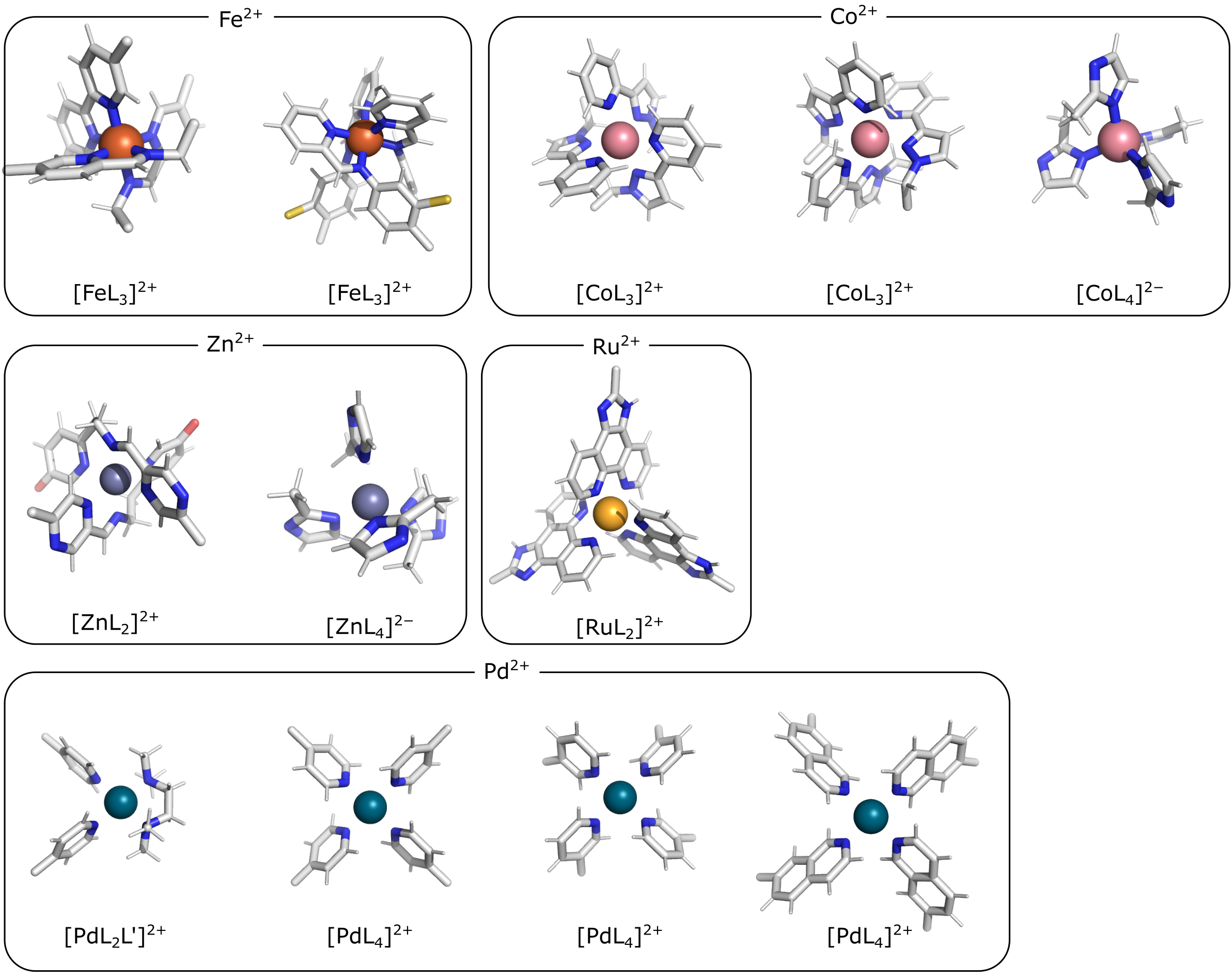
Initial templates available as part of metallicious.
Lennard-Jones
metallicious overwrites metal parameters using Lennard-Jones (LJ) parameters taken from literature. In particular, it is possible to use listed below parameters:
merz-OPC [Merzopc]
merz-opc3 [Merzopc]
merz-tip3p-fb [Merzopc]
merz-tip4p-fb [Merzopc]
merz-spce [Merztip3p]
merz-tip3p [Merztip3p]
merz-tip4-ew [Merztip3p]
zhang-tip3p [zhang]
zhang-opc3 [zhang]
zhang-spce [zhang]
zhang-spceb [zhang]
zhang-tip3p-fb [zhang]
zhang-opc [zhang]
zhang-tip4p2005 [zhang]
zhang-tip4p-d [zhang]
zhang-tip4p-fb [zhang]
zhang-tip4p-ew [zhang]
uff [uff]
Periodic table below shows for which elements L-J parameters are available.

Available L-J parameters in metallicious. L-J parameters for most of the elements are available from UFF [uff]. L-J parameters for some of the metals were derived by Merz et al. [Merzopc] [Merztip3p] and Zhang et al. [zhang] to reproduce hydration free energies and coordination number in aqueous complex.
References:
(a) Monovalent: Sengupta, A.; Li, Z.; Song, L. F.; Li, P.; Merz, K. M. Parameterization of Monovalent Ions for the OPC3, OPC, TIP3P-FB, and TIP4P-FB Water Models. J. Chem. Inf. Model. 2021, 61 (2), 869–880. https://doi.org/10.1021/acs.jcim.0c01390, (b) Divalent: Li, Z.; Song, L. F.; Li, P.; Merz, K. M. Systematic Parametrization of Divalent Metal Ions for the OPC3, OPC, TIP3P-FB, and TIP4P-FB Water Models. J. Chem. Theory Comput. 2020, 16 (7), 4429–4442. https://doi.org/10.1021/acs.jctc.0c00194. (c) Tri- and Tetravalent: Li, Z.; Song, L. F.; Li, P.; Merz, K. M. Parametrization of Trivalent and Tetravalent Metal Ions for the OPC3, OPC, TIP3P-FB, and TIP4P-FB Water Models. J. Chem. Theory Comput. 2021, 17 (4), 2342–2354. https://doi.org/10.1021/acs.jctc.0c01320.
(a) Monovalent: Li, P.; Song, L. F.; Merz, K. M. Systematic Parameterization of Monovalent Ions Employing the Nonbonded Model. J. Chem. Theory Comput. 2015, 11 (4), 1645–1657. https://doi.org/10.1021/ct500918t. (b) Divalent: Li, P.; Roberts, B. P.; Chakravorty, D. K.; Merz, K. M. Rational Design of Particle Mesh Ewald Compatible Lennard-Jones Parameters for +2 Metal Cations in Explicit Solvent. J. Chem. Theory Comput. 2013, 9 (6), 2733–2748. https://doi.org/10.1021/ct400146w. (c) Tri- and Tetravalent: Li, P.; Song, L. F.; Merz, K. M. Parameterization of Highly Charged Metal Ions Using the 12-6-4 LJ-Type Nonbonded Model in Explicit Water. J. Phys. Chem. B 2015, 119 (3), 883–895. https://doi.org/10.1021/jp505875v.
(a) Monovalent: Qiu, Y.; Jiang, Y.; Zhang, Y.; Zhang, H. Rational Design of Nonbonded Point Charge Models for Monovalent Ions with Lennard-Jones 12–6 Potential. J. Phys. Chem. B 2021, 125 (49), 13502–13518. https://doi.org/10.1021/acs.jpcb.1c09103. (b) Divalent: Zhang, Y.; Jiang, Y.; Peng, J.; Zhang, H. Rational Design of Nonbonded Point Charge Models for Divalent Metal Cations with Lennard-Jones 12-6 Potential. J. Chem. Inf. Model. 2021, 61 (8), 4031–4044. https://doi.org/10.1021/acs.jcim.1c00580. (c) Tri- and Tetravalent: Zhang, Y.; Jiang, Y.; Qiu, Y.; Zhang, H. Rational Design of Nonbonded Point Charge Models for Highly Charged Metal Cations with Lennard-Jones 12-6 Potential. J. Chem. Inf. Model. 2021. https://doi.org/10.1021/acs.jcim.1c00723.
Rappé, A. K.; Casewit, C. J.; Colwell, K. S.; Goddard, W. A.; Skiff, W. M. UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114 (25), 10024–10035. https://doi.org/10.1021/ja00051a040.